


De l’importance d’anticiper la sécurité biologique des DM dès la conception
La maîtrise de la sécurité biologique d'un dispositif médical est l'une des exigences essentielles de sécurité et de performance du RDM. CEHTRA prône une approche systématique à adopter dès les premières étapes du développement du produit, avec un maître-mot : anticipation.

Imen Hamdouni (crédit photo : Virginie Lechene)
Par Imen Hamdouni et Paul Fernandes, toxicologues chez CEHTRA
En tant que fabricant, anticiper la sécurité biologique de son dispositif médical (DM) dès la conception est primordial pour se conformer aux exigences du règlement européen (RDM 2017/745). En effet, l'évaluation de la sécurité biologique doit être planifiée de manière rigoureuse, comme le recommande la norme EN ISO 10993-1, qui est le référentiel pour l’évaluation biologique des DM.
Dès les premières étapes de la conception, il est essentiel de caractériser les matériaux afin de comprendre leurs propriétés et leurs effets potentiels sur la santé humaine pour l’utilisation revendiquée par le fabricant du DM.
Une planification adéquate peut minimiser l'étendue des tests nécessaires en identifiant les risques potentiels et en mettant en œuvre des stratégies de mitigation appropriées.

Paul Fernandes (crédit photo : CEHTRA)
L'approche de "case à cocher" doit absolument être évitée, car elle peut conduire à une évaluation superficielle et incomplète des risques biologiques. Pour chaque effet biologique identifié, une justification scientifique solide doit être fournie.
L'interprétation de l'évaluation globale de la biocompatibilité doit également être intégrée dans le cadre du concept de bénéfice-risque, afin d'assurer que les avantages du DM l'emportent sur ses risques potentiels. L’évaluation du risque biologique doit être réalisée en lien avec l’analyse de risques du DM, qui est initiée dès le début de la conception.
Biocompatibilité et EGSP
Le règlement européen 2017/745 exige que le fabricant légal d’un DM réponde à des Exigences Générales de Sécurité et de Performance (EGSP). Il doit notamment apporter la preuve que le DM final est sûr et efficace, pour les patients ou utilisateurs, en conditions normales d'utilisation, tout au long de son cycle de vie.
La documentation technique fournie par le fabricant doit démontrer la conformité aux EGSP, notamment aux exigences 10.1, 10.2, 10.3 et 10.4, qui concernent spécifiquement la sécurité biologique. Pour répondre à ces exigences, il est fortement recommandé d'utiliser la série de normes ISO 10993 harmonisées, ou en voie de l’être, qui fournit les lignes directrices pour l'évaluation de la biocompatibilité des matériaux.
Un DM conforme aux normes harmonisées applicables, ou à des parties pertinentes de ces normes, dont les références ont été publiées au Journal officiel de l'Union européenne, est présumé conforme aux exigences du RDM 2017/745, ce qui favorise sa mise sur le marché.

Sélectionner les matériaux appropriés, c’est choisir les fournisseurs adéquats, en s'assurant que tout changement fera l'objet d'une notification proactive (crédit photo : Crédit photo : © NanzXy; © atipong; © AS Photo Family - stock.adobe.com / Généré à l’aide de l’IA_[M]-CEHTRA).
Dès la conception du DM, il est crucial d'anticiper sa catégorisation selon la norme EN ISO 10993-1. Cela implique de définir son utilisation, les populations cibles, la durée de contact cumulée, et la nature du contact pour chaque partie du dispositif dans le cas où ce dernier est composé de différentes parties ayant des contacts ou des utilisations différentes.
Cette catégorisation permettra d'orienter vers les effets biologiques à maîtriser, les tests nécessaires et d'identifier les matériaux appropriés.
Choisir les matières premières
Le choix des matériaux est une étape clé dans la conception des DM. Il est important de sélectionner des matériaux dont la composition est connue, les spécifications fixes et dont les caractéristiques et performances ne sont pas altérées pendant le processus de fabrication jusqu’au transport et au stockage du DM final. Un certificat d’analyse à jour doit être fourni pour chaque matériau utilisé.
Les matériaux doivent être choisis selon leur compatibilité avec les tissus, les cellules et les liquides biologiques. La compatibilité entre les différents matériaux utilisés pour la fabrication est également primordiale et doit être vérifiée afin d’éviter toute interaction ou altération pouvant générer de nouveaux risques biologiques.
Les matériaux d'emballage, les substances relarguées lors de l'utilisation et les produits de dégradation prévus ou prévisibles doivent aussi être évalués pour garantir leur biocompatibilité.
Enfin, le choix de matériaux pertinents permet de minimiser les risques liés aux contaminants, aux résidus et à la dégradation, ainsi que de réduire au minimum les risques liés aux substances cancérigènes, mutagènes et toxiques pour la reproduction (CMR), et aux perturbateurs endocriniens.
Les fabricants de DM sont naturellement amenés à favoriser des matériaux revendiqués comme biocompatibles. Si le matériau a déjà été testé, il est important de vérifier l’adéquation et la fiabilité des tests réalisés et de s’assurer que les résultats des essais sont toujours valables par rapport à la version actuelle des normes ISO (Etat de l’art).
Cela implique notamment la maîtrise de la différence entre une conformité revendiquée aux classes établies par la Pharmacopée des États-Unis (USP) et une conformité revendiquée à la série de normes ISO 10993. Il convient aussi de procéder à la revue critique des certificats fournis attestant des tests réalisés, des référentiels appliqués, de la description détaillée des éléments testés qui doivent être représentatifs du dispositif médical final, et des résultats obtenus.
Bien que l'utilisation de matériaux biocompatibles soit un bon point de départ, cela ne garantit pas que le DM final sera également biocompatible. Des étapes de fabrication, d'emballage, de stérilisation et de stockage peuvent avoir un impact significatif sur la biocompatibilité. Tout comme les matériaux, additifs, contaminants et résidus de processus de fabrication doivent être identifiés et qualifiés.
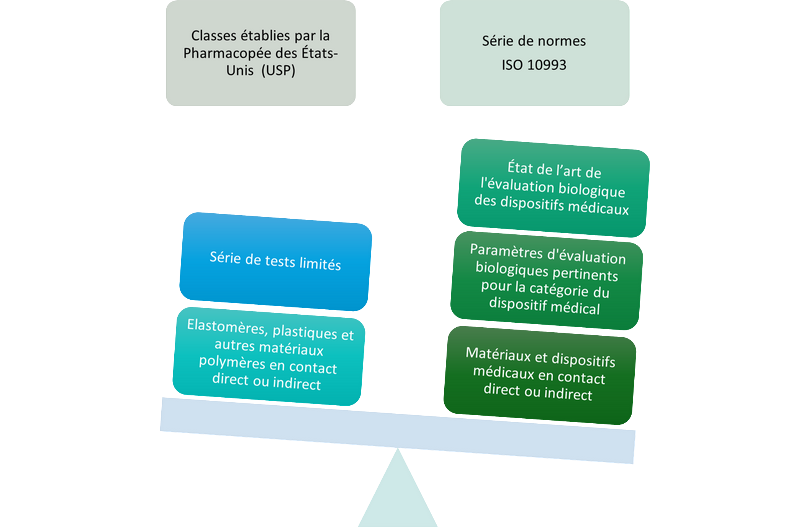
Il faut savoir distinguer une conformité revendiquée aux classes établies par la Pharmacopée des États-Unis (USP) d'une conformité revendiquée à la série de normes ISO 10993 (crédit photo :
CEHTRA).
Choisir le/les fournisseur(s) de(s) matière(s) première(s)
Cela apparaît comme une évidence : sélectionner les matériaux appropriés, c’est choisir les fournisseurs adéquats. Anticiper le nombre de fournisseurs pour une même matière première et privilégier ceux qui disposent d'un système de management de la qualité en place semble une recommandation pertinente. La notification proactive des changements effectués par le fournisseur dans la chaîne de production est nécessaire.
Les fournisseurs doivent également garantir la fiabilité des livraisons et la disponibilité des produits dans les délais prévus, tout en offrant des prix compétitifs.
En conclusion...
CEHTRA met son expertise en évaluation du risque biologique au service des fabricants pour les accompagner, de la conception jusqu’au suivi post-commercialisation, en passant par le marquage CE.
La maîtrise de la sécurité biologique des DM est un processus complexe qui nécessite une planification rigoureuse et une approche systématique à adopter dès les premières étapes du développement du DM.
En intégrant les exigences réglementaires, en choisissant judicieusement les matériaux et les fournisseurs, et en anticipant les besoins en biocompatibilité dès la conception, les fabricants peuvent garantir que leurs dispositifs médicaux sont sûrs et efficaces tout au long de leur cycle de vie.
En planifiant et en documentant ces aspects dès la conception du dispositif médical, tout en maintenant une mise à jour continue de l'évaluation biologique pour répondre aux exigences réglementaires, les fabricants évitent des retards coûteux, réduisent les risques de non-conformité et assurent la sécurité des patients et des utilisateurs.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn



