


Faciliter l’accès aux marchés européen et U.S. des DM logiciels de classe II

Des élèves de l'Université de Technologie de Compiègne (UTC) ont élaboré, dans le cadre d'un mémoire de Master, un nouveau guide très utile. Son but : aider les éditeurs de DM logiciels de classe II à préparer la mise en conformité de leurs produits pour accéder aux marchés européen et américain.

(source photos : UTC)
Par Manal Bouramdane, Kevine Dzegang, Haitam Kadiri, Hoda Slika, Charles Yanze, encadrés par Julie Follet de l'UTC
Le marché des dispositifs médicaux (DM) logiciels connaît une expansion significative, portée par l'émergence de nouvelles pathologies, les avancées technologiques et la numérisation croissante du secteur de la santé (D. Valdes, 2022). Il s’agit majoritairement de dispositifs de classe II. Fin 2024, ils représentaient 65 % des DM commercialisés en Europe référencés sur la base de données EUDAMED, et 43 % des DM commercialisés outre-Atlantique référencés sur la base de données GUDID. Leur mise sur le marché reste complexe car impactée par des règlementations hétérogènes entre l’Europe et les États-Unis. De fait, les petites et moyennes entreprises font face à des difficultés importantes liées aux délais et aux coûts de mise en conformité règlementaire.
Un guide pratique pour répondre aux exigences des deux marchés
C’est pour les accompagner à chaque étape clé de la mise en conformité de leurs produits, qu'a été élaboré ce guide (disponible gratuitement ici).
Tout d’abord, la classification du DM consiste à identifier précisément la classe de risque du dispositif selon les critères règlementaires en vigueur. Ensuite, pour la préparation du dossier technique, le guide renvoie les utilisateurs vers les recommandations applicables pour une documentation claire, complète et conforme aux exigences réglementaires. L'utilisateur se voit aussi présenter l'obtention de l’identifiant unique des dispositifs (UDI), garante d’une traçabilité optimale des produits et fournisseurs dans les bases de données de référence. Enfin, le guide met l'accent sur la surveillance post-commercialisation, une phase critique pour évaluer les performances des dispositifs et signaler rapidement les éventuels incidents, contribuant ainsi à maintenir les standards de sécurité et de qualité.
Ce guide se distingue par son approche simplifiée et adaptable. Il offre une présentation détaillée des normes applicables en Europe et aux Etats-Unis et des outils téléchargeables pour aider à structurer les dossiers techniques, répondre efficacement aux audits et garantir une conformité continue.
Quelles convergences et divergences entre les deux réglementations ?
Les DM logiciels de risque modéré pour les patients nécessitent malgré tout des évaluations rigoureuses avant leur mise sur le marché. Le tableau affiché plus bas synthétise les similitudes et différences entre les approches règlementaires aux États-Unis (Titre 21 du Code of Federal Regulations) et en Europe (Règlement (UE) 2017/745). Il prend notamment en compte les exigences spécifiques de mise sur le marché et les processus de surveillance post-commercialisation dans chaque région.
Cette représentation dresse une vue d’ensemble claire et concise, facilitant ainsi la compréhension des principaux points à considérer pour les entreprises. Les écarts entre les réglementations américaine et européenne s’expliquent par des différences fondamentales d’approche de mise sur le marché. En Europe, les entreprises doivent obtenir le marquage CE médical, un processus pour lequel le délai d'approbation du certificat varie généralement entre 12 et 18 mois. Aux États-Unis, les procédures sont plus rapides, allant de 3 mois pour les dispositifs suivant le principe d’équivalence (ou prédicat, selon la procédure 510(k)) à 4 mois pour les dispositifs suivant la procédure De Novo (sans dispositif équivalent déjà commercialisé sur le marché américain). Les coûts d’approbation sont également différents : environ 35 k€ en Europe, contre 6 k€ à 15 k€ aux États-Unis (G. Promé, 2022).
Ces différences soulignent l’importance d’une préparation adaptée pour naviguer efficacement dans ces deux environnements réglementaires.
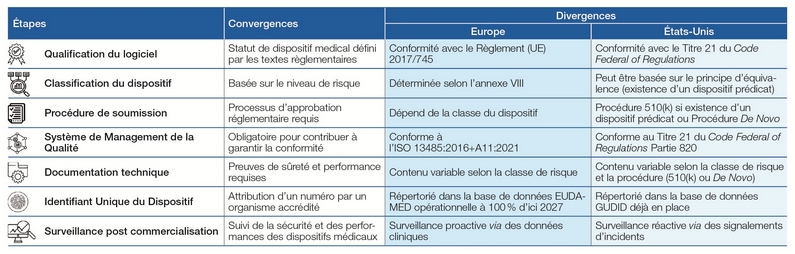
Analyse comparée des exigences règlementaires propres à la mise sur le marché des dispositifs médicaux logiciels de classe II aux États-Unis et en Europe (source : Auteurs).
Perspectives d’amélioration du guide
La mise sur le marché des DM logiciels de classe II aux Etats-Unis et en Europe exige une compréhension fine des règlementations et des étapes administratives en vigueur. Ce guide se veut un outil clé pour les petites entreprises, leur permettant d’assurer une conformité complète de leurs produits tout en optimisant les délais et coûts associés.
Etablissement de formation de référence dans le secteur du DM, l'UTC propose un master en Ingénierie de la Santé (IDS) avec deux parcours : Technologies biomédicales et territoire de santé (TBTS) et Dispositif médical et affaires réglementaires (DMAR).
Des professionnels, notamment responsables qualité, responsables affaires réglementaires et auditeurs, ont souligné l’utilité de cet outil, en proposant des suggestions pour renforcer son efficacité. Il est question, par exemple, d'intégrer un questionnaire interactif pour personnaliser les étapes de mise en conformité restant à suivre.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn



