


Révision de la norme ISO 10993-18 : introduction au concept de l’AET

A l’approche de la parution de la nouvelle révision de la norme ISO 10993-18 sur la caractérisation chimique des matériaux des DM, Albhades se propose de faire le point sur l’analyse des extractibles réalisée dans le cadre des études de biocompatibilité des dispositifs médicaux.

Frédéric Mirguet
Par Frédéric Mirguet, directeur du développement des produits de santé chez Albhades
L’évaluation biologique des dispositifs médicaux est réalisée selon les normes de la série ISO 10993 et repose sur un processus de gestion des risques. Dans sa révision d’août 2018, la norme ISO 10993-1 précise que la collecte d’informations chimiques et physiques est une première étape cruciale pour l’évaluation des dispositifs médicaux en contact direct ou indirect avec le patient. Selon l’exposition clinique et les informations disponibles, la caractérisation physico-chimique pourra nécessiter des efforts plus ou moins importants.
La première étape de la caractérisation chimique consiste à compiler l’ensemble des informations disponibles telles que les matériaux constitutifs du dispositif médical, les données de process, les matériaux d’emballage ou les résidus de stérilisation (si pertinent) qui peuvent tous, potentiellement, conduire à la présence de résidus chimiques susceptibles d’impacter la santé du patient et/ou la qualité du dispositif médical.
Dans la plupart des cas, la compilation des données disponibles ne suffit pas à caractériser le risque chimique parce qu’il n’est pas possible d’estimer la concentration réelle de tel ou tel résidu sur le dispositif médical et parce que toutes les données ne sont pas forcément disponibles (produits chimiques dont la formule est protégée par un brevet, produits de dégradation, interactions chimiques entre les résidus et leurs éventuels produits de dégradation…). Pour ces différentes raisons, il est nécessaire de réaliser une étude de caractérisation chimique selon la norme ISO 10993-18 (également appelée étude "extractibles") puis d’évaluer toxicologiquement les résultats obtenus selon la norme ISO 10993-17.
Traduire une limite toxicologique en limite analytique
A cet égard, la nouvelle version de la norme ISO 10993-18 permet de mieux prendre en considération le risque chimique en introduisant le concept d’AET (Analytical Evaluation Threshold ou seuil d’évaluation analytique) pour l’évaluation des composés organiques. Déjà utilisé depuis plusieurs années dans l’industrie pharmaceutique, ce concept a été adapté au cas des dispositifs médicaux et permet de traduire une limite toxicologique (en µg/dispositif médical ou en µg/jour) en limite analytique (µg/mL) directement exploitable par le chimiste.
L’AET peut être calculé comme suit :
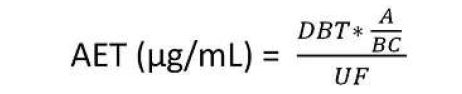
Dans cette équation, DBT est la limite toxicologique la plus critique à prendre en considération pour l’étude ("Dose Based Threshold"). Le DBT peut être assimilé au TTC ("Threshold of Toxicological Concern") ou au SCT ("Safety Concern Threshold"). Ces valeurs sont données par la littérature en µg/jour et il convient de les convertir en µg/dispositif.
Quoi de neuf chez Albhades ?
Albhades a franchi bon nombre d'étapes ces derniers mois :
- Passage avec succès de l’audit de suivi pour les référentiels Certipharm, ISO 9001 et ISO 13485 et de l’audit de renouvellement de l’accréditation Cofrac selon l’ISO 17025 v2017
- Passage avec succès de l’audit pour la demande d’extension d’accréditation Cofrac pour la validation de nettoyage de DM.
- Enregistrement auprès de la FDA du groupe et de sa plateforme d’analyses structurales. Cet enregistrement présente un avantage important pour les sociétés présentes sur le marché US.
- Avec une croissance du chiffre d’affaires de 30 % ces deux dernières années, le groupe recrute et est sur le point de passer la barre des 100 employés (50 il y a 3 ans)
- Investissement : le groupe complète son offre packaging en proposant le bubble test (ASTM F2096) et en s’équipant d’un dynamomètre de dernière génération, apte à réaliser des essais selon la dernière version de la norme NF EN 868-5. Ces investissements complètent les derniers effectués : 1 ILC, 2 HS-GC/FID, 1 HS-GC/MS …
- Nouvelles prestations d’identification microbiologique : protéotypique & génotypique
- Enregistrement des échantillons : en plus des aménagements importants prévus, le groupe accueillera un tout nouvel outil d’enregistrement des échantillons à l’horizon 2020. Ce logiciel permettra un meilleur service au client.
Par ailleurs, A est le nombre de dispositifs médicaux à extraire ; B est le volume d’extraction (mL) ; C est le coefficient relatif à l’exposition clinique correspondant au ratio entre la surface en contact avec le patient et la surface extraite.
Les méthodes analytiques utilisées pour l’évaluation des extractibles sont destinées à rechercher des composés inconnus (screening) et doivent donc détecter le maximum de composés. Au contraire d’un dosage ciblé, ces méthodes utilisent des détecteurs capables d’identifier les composés inconnus (spectromètres de masse) et les quantifications sont réalisées selon des composés standards de substitution conduisant à des niveaux de précision inférieurs à ceux des méthodes utilisées pour des dosages ciblés. L’incertitude qui en découle est prise en compte par le facteur UF de l’équation. L’utilisation de plusieurs types de détecteurs (UV, diffusion de la lumière …) et la multiplication du nombre de standards de substitution permettent de limiter cette incertitude.
Pour que l’analyse soit conforme à l’objectif attendu, l’AET doit être supérieur à la limite de quantification de la méthode (LOQ) calculée selon le standard de substitution ayant le signal le plus faible et vérifiée lors de chaque étude. Dans ce contexte, les ratios d’extraction décrits dans la norme ISO 10993-12 : 2012 ne sont donc plus vraiment adaptés pour la caractérisation chimique si les conditions de l’extraction sont établies pour ne pas dégrader le dispositif médical. Néanmoins, cette norme étant toujours en vigueur, il conviendra de justifier tout écart.
Choix de la limite toxicologique : des principes inspirés de la pharma
Le choix de la limite toxicologique (DBT) est particulièrement critique pour la réalisation de l’étude. Usuellement, les composés mutagènes sont considérés comme les plus à risque. La ligne directrice ICH M7 applicable dans l’industrie pharmaceutique fixe des limites pour ces composés. Ainsi, pour la durée d’exposition la plus longue, la valeur de 1,5 µg/jour est inférieure à la valeur seuil attribuée à la classe de Cramer la plus conservatrice et la plus protectrice pour des composés non cancérigènes (90 µg/jour). Par conséquent, la valeur de 1,5 µg/jour est présumée très protectrice vis-à-vis des effets cancérogènes et non cancérogènes pour un médicament. Ces principes ont été adaptés aux dispositifs médicaux dans la spécification technique ISO/TS 21726:2019 qui précise également les valeurs à prendre en compte en fonction de la durée de contact du dispositif avec le patient.
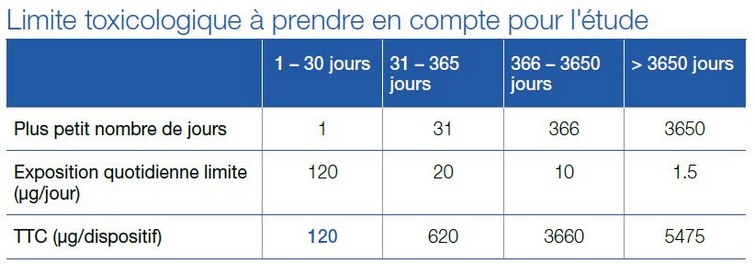
Ces documents donnent des valeurs en µg/jour et non en µg/dispositif et il convient donc de ne pas faire l’amalgame entre ces unités. Par exemple, les valeurs indiquées dans ICH M7 sont applicables à des médicaments et tiennent compte de prises répétées, ce qui n’est pas applicable aux dispositifs médicaux pour lesquels le risque patient dépend d’une cinétique de relargage. La cinétique étant rarement connue, l’étude des extractibles dans les dispositifs médicaux long terme doit être réalisée selon des conditions d’extraction exhaustives, ce qui permet alors de considérer la valeur de 120 µg/dispositif comme étant la plus protectrice pour le patient, tel que démontré par le tableau ci-dessous.
Le concept d’AET n’est pas applicable aux composés appelés "cohort of concern" présentant un risque toxicologique spécifique non proportionnel à la dose. De même, l’analyse élémentaire suppose l’application de limites toxicologiques propres à chaque élément (voir par exemple ICH Q3D).
Le cadre de l’étude étant posé, tout composé présentant une valeur inférieure à l’AET sera considéré comme non à risque pour le patient.
Dans le cas contraire, il convient de réaliser une étude toxicologique formelle selon la norme ISO 10993-17. L’AET ayant été fixé sur la base d’une limite pour un composé mutagène, chaque composé présentant une concentration supérieure à l’AET sera passé en revue pour déterminer le risque toxicologique en fonction de sa propre limite de toxicité.
En conclusion, la nouvelle norme ISO 10993-18 constitue un réel progrès pour les laboratoires qui peuvent désormais établir des stratégies analytiques réellement adaptées au contexte clinique et toxicologique d’une étude.
 Twitter
Twitter LinkedIn
LinkedIn





