


Essais cliniques de prototypes de DM : maîtriser la conformité réglementaire
Les instituts de recherche ou les start-up disposent rarement d'une organisation qui permette le suivi réglementaire de leurs activités de conception et de production. Surgiqual Institute les accompagne dans cette démarche en leur apportant son expertise réglementaire et ses processus de conception certifiés ISO 13485.

Lucile Marron Brignone, Ph. D, Directrice Qualité & Affaires Réglementaires
Le renforcement de la réglementation encadrant les dispositifs médicaux (DM) et DM de diagnostic in vitro transparaît dans les nouveaux règlements (UE) 2017/745 et (UE) 2017/746 qui intègrent de nouvelles exigences d’essais cliniques pour démontrer les performances attendues.
Ce constat se reflète entre autres dans :
- le durcissement des classes de risques : citons ici l'exemple des implants actifs ou celui de nombreux implants chirurgicaux et prothèses, qui appartiennent désormais d’office à la classe III. Autres DM concernés : les logiciels médicaux intervenant dans les décisions thérapeutiques ou diagnostiques affectés au minimum à la classe IIa.
- des exigences générales de sécurité et de performances plus détaillées et plus nombreuses,
- une procédure d’évaluation clinique imposée pour apporter la preuve clinique de la performance médicale ou technique revendiquée.
Plus l’innovation sera importante, plus les essais cliniques seront indispensables, surtout en l’absence d’équivalence et/ou de cadre normatif prédéfini. L’obtention de preuves cliniques est par ailleurs une étape clé pour les instituts de recherche et start-up dans la valorisation, la recherche de financement et/ou l’accès au marché.
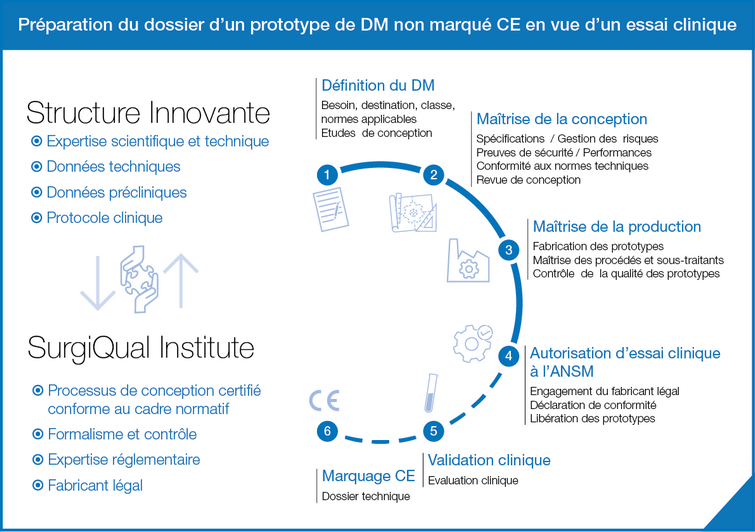
Que le fabricant soit un industriel déjà présent sur le marché, un institut de recherche ou encore une start-up, il aura les mêmes obligations concernant les prototypes de DM non marqués CE, notamment assurer la sécurité des patients. Dans le cadre d’un dossier de demande d’autorisation à l’ANSM, il s’agit pour le fabricant de démontrer :
- la pertinence du suivi de sa conception au regard des exigences de la norme ISO 13485 (plus l’IEC 62304 pour les logiciels) et des normes particulières applicables ;
- l’évaluation des risques selon la norme ISO 14971 en relation avec la démonstration argumentée d’un bénéfice médical attendu ;
- la maîtrise des processus de fabrication des prototypes destinés aux essais cliniques et ce, avec des éléments de validation plus nombreux lorsque la classe de risque augmente.
Sur la base de ces éléments, le fabricant doit : (1) Produire une déclaration de conformité avec une évaluation faite de la réponse aux exigences applicables de l’annexe I des nouveaux règlements DM ou DMDIV, (2) Produire un certificat de libération des prototypes après vérification que tous les critères d’acceptation définis sont atteints, (3) Évaluer le risque résiduel final pour les utilisateurs du dispositif dans le cadre défini de l’essai.
Or, les instituts de recherche ou start-up disposent rarement d'une organisation permettant le suivi des activités de conception et production (généralement un système qualité ISO 13485), et peuvent difficilement engager leur responsabilité sur des expertises qu’ils ne maîtrisent pas ou pas encore.
Ils peuvent bien sûr internaliser une organisation qualifiée, de conception, contrôle et certifications produits, mais c’est une démarche longue et coûteuse et qui peut allonger les délais de valorisation.
C’est pour éviter ces écueils que Surgiqual Institute (SQI) a mis en place un processus d’accompagnement ciblé. Il associe à l’expertise technique de ses clients, son expertise réglementaire et ses processus de conception certifiés ISO 13485 :
- les données techniques de la conception sont redéployées dans un processus de conception conforme aux exigences de l’ISO 13485 ;
- les preuves et les éléments de sécurité/performance/conformité nécessaires sont déterminés puis vérifiés et tracés ;
- les activités de production de la structure et de ses sous-traitants pour l’obtention des prototypes sont mises sous contrôle.
Ce dossier répondant aux exigences réglementaires de la conception du DM/DMIA/DMDIV restera la propriété du client qui pourra l’intégrer dans son propre système qualité le moment venu.
 Twitter
Twitter LinkedIn
LinkedIn





