


Cybersécurité : de nouvelles exigences pour les fabricants de DM connectés

Avec l'essor des dispositifs médicaux connectés et la multiplication des cas de vulnérabilité avérés, la cybersécurité est devenu un enjeu majeur. Cyrille Michaud nous explique les exigences qu'impose la réglementation à ce sujet, et comment cela se traduit pour les fabricants concernés.
 Par Cyrille Michaud, consultant en dispositifs médicaux logiciels, MD101 Consulting
Par Cyrille Michaud, consultant en dispositifs médicaux logiciels, MD101 Consulting
Si le terme remonte aux années 80, la cybersécurité est un sujet très récent dans le monde des dispositifs médicaux. Avant les années 2010 et l'apparition de logiciels DM à part entière ou de DM connectés, personne ne se souciait de cybersécurité. Mais tout a changé depuis quelques années, notamment à la lumière de nombreux cas de piratage qui ont fait les gros titres. La cybersécurité est désormais un véritable enjeu pour un nombre croissant de fabricants. Car ceux qui n'auraient pas déjà mis sur le marché un logiciel ou un dispositif connecté, sont nombreux à vouloir le faire.
La principale caractéristique des dispositifs connectés est leur capacité à communiquer des données personnelles de santé. Ces données peuvent être utilisées par exemple pour de l'aide au diagnostic, ou du suivi de patient.
De ce fait, les réglementations applicables aux dispositifs connectés incluent la réglementation des dispositifs médicaux, mais aussi la réglementation sur les données personnelles.
Pour les deux marchés qui concernent le plus les fabricants de DM européens, les réglementations applicables sont celles de l’Union Européenne et des Etats-Unis.
Les réglementations aux Etats-Unis
Outre-Atlantique, le contexte est relativement simple comparé à celui de l'Europe. Il y a deux réglementations applicables aux Etats-Unis :
- le 21 CFR, principalement la partie 820 pour le système d’assurance qualité, et la partie 11 pour les enregistrements et signatures électroniques,
- les Health Insurance Portability and Accountability Act (HIPAA) et Health Information Technology for Economic and Clinical Health Act (HITECH), deux textes qui traitent de nombreux sujets et portent, dans le cas qui nous intéresse, sur la protection et la confidentialité des données de santé.
La réglementation HIPAA/HITECH n'est pas directement applicable aux fabricants de dispositifs médicaux mais à des entités concernées (covered entities), par exemple des hôpitaux ou des compagnies d'assurance, qui font usage de DM connectés. Les fabricants de ces dispositifs doivent donc satisfaire aux contraintes de l'HIPAA/HITECH pour que leurs produits puissent être utilisés par les entités concernées.
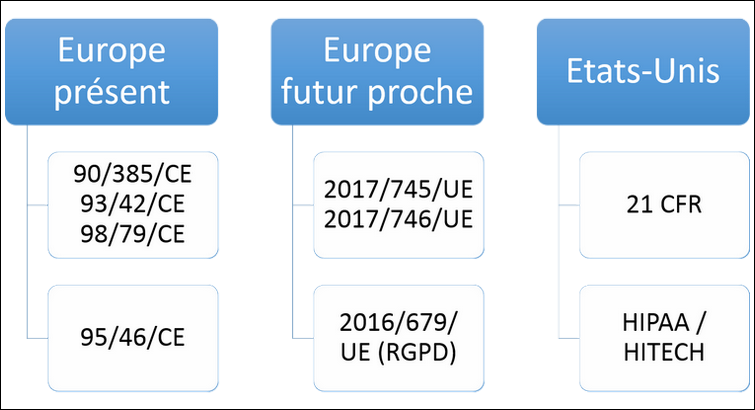
Réglementations applicables aux DM connectés aux États-Unis et en Europe.
Les réglementations en Europe
Les réglementations applicables en Europe sont les directives 93/42/CE, 90/385/CE et 98/79/CE sur les dispositifs médicaux et la directive 95/46/CE sur la protection des données.
Les trois directives sur les dispositifs médicaux sont totalement muettes en matière de cybersécurité. Il n'existe pas non plus de guide MEDDEV ou autre sur la cybersécurité.
La directive sur les données personnelles contient des exigences sur la confidentialité et la sécurité des données, sans être aussi précises que les exigences états-uniennes.
Mais face à l’évolution de la technologie, ces directives sont proches de leur fin de vie et vont être remplacées par les règlements 2017/745 et 2017/746 (sur les DM et DM DIV) et le règlement 2016/679 (RGPD pour "Règlement Général sur la Protection des Données").
Les règlements sur les dispositifs médicaux, qui seront applicables respectivement en 2020 et 2022, introduisent de nouvelles exigences de sécurité. Les exigences incluent des rubriques sur l'environnement informatique, l'interopérabilité et la sécurité de l'information. Il ne s'agit que de quelques phrases, au regard du volume de ces textes réglementaires très touffus. Mais il ne faut pas s'y tromper, c'est un changement majeur comparé aux directives.
Un règlement sur la protection des données applicable en mai 2018
Le RGPD est encore plus contraignant sur les données personnelles, avec de très nombreuses exigences, en particulier sur les données de santé personnelles, comme le droit d'accéder facilement à ses données, le droit à l'oubli, l'obligation pour les opérateurs de conduire une étude d'impact sur la vie privée (PIA pour Privacy Impact Assessment) et l'obligation de s'assurer de la sécurité par la conception.
Le RGPD se traduit par une charge de travail considérable pour tout organisme qui manipule des données personnelles et en particulier des données de santé. Ce règlement sera totalement applicable en mai 2018, il reste donc peu de temps aux fabricants de dispositifs pour l'appliquer.
A noter que la Commission Nationale Informatique et Libertés (CNIL) a rédigé un guide : "Règlement européen : se préparer en 6 étapes", disponible depuis la page d'accueil du site www.cnil.fr.
De même que la réglementation HIPAA/HITECH, le RGPD n'est pas directement applicable aux fabricants de dispositifs médicaux, mais aux organismes qui traitent des données de santé au moyen de ces dispositifs. Les dispositifs médicaux doivent donc être conçus pour pouvoir respecter les exigences du RGPD. Ce qui demande d'y passer du temps. Car mettre en place, dans une base de données, un moyen efficace pour permettre d'effacer totalement les informations d’une personne, afin de respecter le droit à l'oubli, n'est pas une tâche facile !
Et la France dans tout ça ?
La France se distingue par l'adoption depuis 1978 d'une réglementation très restrictive sur les données personnelles, incarnée par la CNIL. A cela, il faut ajouter les textes de loi sur l'hébergement des données de santé.
Le caractère très restrictif de la loi française rend paradoxalement les choses très simples. Un organisme qui veut stocker des données de santé doit les stocker chez un Hébergeur Agréé de Données de Santé (HADS). La liste de ces HADS est disponible sur le site de l'ASIP Santé : esante.gouv.fr.
Même si la loi française est applicable de longue date, il reste nécessaire d'effectuer une analyse d'écart avec le RGPD car certaines exigences du RGPD n'existent pas dans la loi française.
UL 2900 : une nouvelle norme reconnue par la FDA
Le 5 juillet dernier a été publié le premier volet d’une série de normes (UL 2900) consacrée à "la cybersécurité des dispositifs médicaux connectables à un réseau". Le but est de fournir aux fabricants des critères mesurables et testables pour évaluer les vulnérabilités d'un logiciel de DM et identifier les améliorations à apporter en matière de cybersécurité.
L’UL 2900-1, qui concerne les exigences générales, sera suivie de l’UL 2900-2-1 (exigences particulière pour les systèmes de santé), de l’UL 2900-2-2 (exigences particulière pour les systèmes de contrôle industriels) et de l’UL 2900-3 (exigences générales relatives aux processus organisationnels d’évaluation des risques pour déterminer les cyber-menaces applicables).
Fin août, la FDA a annoncé l’adoption de l’UL 2900-1, ainsi que 8 autres normes relative à l’informatique de santé, dont l’IEC 82304-1 sur les logiciels de santé.
Des exigences lourdes de conséquences pour les fabricants
En résumé, la réglementation exige que la conception des dispositifs médicaux prenne en compte la sécurisation du produit et des données traitées, que les données soient stockées chez un hébergeur agréé, et qu’elles puissent être traitées selon un ensemble de règles telles que le droit à l’oubli.
Quelle que soit la réglementation, la mise en œuvre de la cybersécurité dans les DM commence donc avant tout par une conception sécurisée.
Dans l'illustration de la page 18, un fabricant de DM offre une fonction de télésuivi des patients. Les données transitent entre le dispositif et un serveur de données via internet pour être exploitées par du personnel médical. La principale difficulté, ici, est la multiplicité des acteurs permettant d’assurer le service de télésuivi : fournisseur d’une "box" relais entre le dispositif et internet, fournisseur télécom, fournisseur d’hébergement agréé et fournisseur de serveur applicatif. La prise en compte des exigences de sécurité demande la contribution de tous ces acteurs, en particulier par la mise en œuvre de mesures de réduction de risques par la conception.
Cette multiplicité des intervenants est relativement nouvelle dans le monde du logiciel DM. On peut faire la comparaison avec les fabricants de dispositifs physiques qui ont l’habitude de sous-traiter une partie des procédés de fabrication, nettoyage, stérilisation... auprès de sociétés qualifiées. Les contraintes sur les dispositifs connectés vont probablement amener à la mise en place d’un écosystème équivalent pour le traitement et le stockage de données. C’est déjà le cas avec les HADS.
Un autre impact concerne la documentation technique logicielle requise pour démontrer la conformité aux exigences de sécurité des nouveaux règlements. Beaucoup de logiciels de DM connectés présentent peu de risques pour les patients. La perte de données médicales ou de connexion ne conduit pas à une dégradation de l’état des patients, qui disposent, en cas de panne, de moyens de rechange plus classiques comme le téléphone. La documentation requise pour ces logiciels est donc succincte. Mais en ajoutant la prise en compte des exigences liées à la cybersécurité, la documentation devient tout de suite plus volumineuse car il faut démontrer la sécurité par la conception.
La documentation logicielle doit alors inclure des spécifications techniques, des diagrammes d’architecture, des protocoles et des rapports de tests fournissant la preuve de la prise en compte des exigences réglementaires de cybersécurité.
AAMI TIR57:2016 : un guide précieux
Publié par l’association américaine AAMI, et reconnu par la FDA, le TIR57:2016 ("Principles for medical device security - Risk management") est un guide fort utile pour les fabricants de DM connectés. C’est d'ailleurs le seul document qui traite de l'application des principes de cybersécurité et de leur impact sur le processus de gestion des risques ISO 14971.
Des coûts supplémentaires à prévoir
Le coût de la conception des logiciels répondant à ces exigences s’en trouve alourdi. Il faut y ajouter le coût d’exploitation avec la location de serveurs d’hébergeurs agréés.
Par ses conséquences sur le cycle de vie d’un dispositif médical, la cybersécurité doit être gérée comme un processus à part entière. A l’instar du processus de gestion des risques patients, les fabricants de dispositifs médicaux doivent inclure un processus de gestion des risques de cybersécurité dans leurs activités. Ce qui implique des ressources allouées, une surveillance et une mesure du processus... et au final des coûts supplémentaires.
 Twitter
Twitter LinkedIn
LinkedIn





